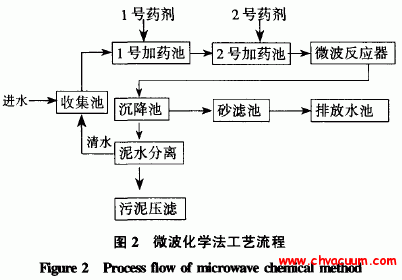
AlN在α-Al2O3(0001)表面吸附过程的理论研究(2)
2、AlN的吸附过程与表面结构
对于模型A 的吸附过程,如图2 所示,图3A ①~ ④所示可分4 个阶段: ①0~0.098ps 为物理吸附过程,最初AlN 分子的Al 朝下被吸附,水平低于N原子约0.03nm ,主要是暴露在基片最外表面O 的电负性大,表面对AlN 分子中的Al 产生了静电弱吸附,这对表面结构产生了明显影响,主要是消除了原有的弛豫,使得a2Al2O3 (0001) 表层Al 暴露在最外表面,由此增加了对AlN 中的N 的静电吸附力,使得AlN 分子的键长由0.1850nm 变为0.2071nm ,从而引起了AlN 分子的键减弱。最后,AlN 分子中的N 距离基底临近Al ②原子的距离为0.2409nm ~0.2544nm ,Al 位于S7 吸附上方, 距表面约为0.200nm~0.072nm ,AlN 分子中的Al 略向[ 1 210 ]方向偏移,其物理吸附能约为0.367eV ,随后,体系能量将发生显著降低。②0.098~0.190ps 为(Al) N -Al ②成键过程。AlN 分子中的N 原子继续向[ 1210 ]方向偏移,并迅速向基底Al ②接近,在0.116ps 是开始(Al ) N —Al ②成键, 键长为0.212nm; 在0.155ps时,形成较强的(Al ) N —Al ②化学键, 键长为0.1797nm ,此时Al 位于S7 吸附位上方,距表面约0.258nm ,吸附能达到2.527eV。③0.190~0.50ps 为Al —O ( 基片) 成键过程。(Al ) N —Al ②键长在0.180nm~0.196nm 之间振动,同时Al 迅速与基片表面O 结合,并略向S6 方向迁移,Al —O (基片) 距离约为0.165nm。④0150ps 后,AlN 分子逐渐同基片表面形成稳定的吸附态。在体系能量最低处(01867ps) ,AlN 分子在a2Al2O3 ( 0001) 表面吸附能为4.844eV ,Al 的吸附位置趋近于S7 (图3A2 ④) 。
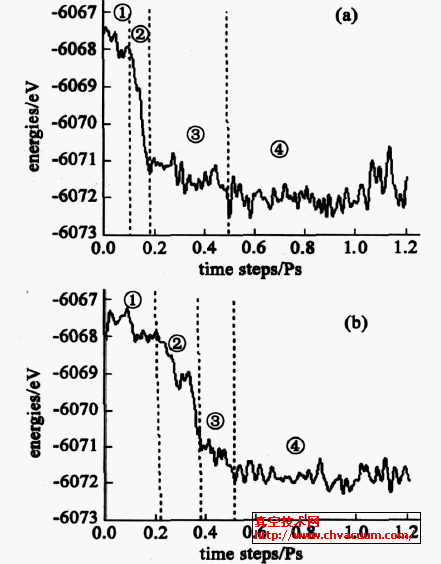
图2 AlN 在模型A 和模型B 中吸附过程与体系能量变化
对于模型B 同模型A 一样,吸附过程与体系能量变化如图2 (b) ,吸附过程也有4 个阶段,如图3 ①~ ④所示: ①0~0.22ps 为表面迁移和物理吸附过程。最初AlN 分子向[10 10 ]迁移,其中Al 朝下被吸附,距表面0.236nm。0.075ps AlN 中的Al 开始向[12 10 ]旋转,AlN 分子中Al 向表面O 接近,Al 向S5 吸附位移动。②0.22~0.38 ps 为(Al) N —Al ②成键过程。同模型A 类似,在0.344ps 时,N —Al ②化学键长为0.162nm ,此时AlN 分子中的Al 位于S5 和S6之间的吸附位上,距离基片表层为0.47nm ,吸附能为3.65eV。③0.38~0.51ps 为Al —O(基片) 成键过程。(Al) N —Al ②保持成键状态,Al 迅速与基片表面的O 结合,位于S5 和S6 之间位置,Al 与表面O距离约为0.32nm ,与相邻的基底O 成键。④0.51ps 之后,随着(Al)N —Al ②、Al —O(基片) 键长键角的振动调整,体系能量下降逐渐趋于稳定。在体系能量最低处(0.97ps) ,AlN 分子在a-A1203 (0001) 表面吸附能为5.132eV ,Al 的吸附位置趋近于S5 (图32B2 ④) 。
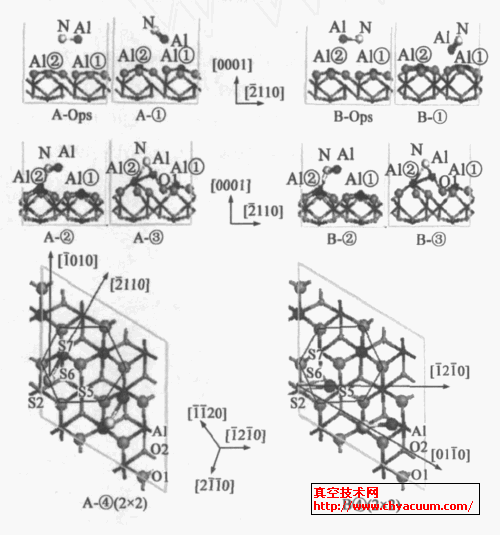
图3 AlN 吸附过程4 个阶段状态轨迹图①物理吸附, ②Al- O(基片) 成键, ③(Al)N - Al 成键, ④稳定的吸附位置。灰色小球代表基底O 原子;黑色小球代表基底Al 原子;白色小球代表N 原子;较大的深灰色小球代表AlN 中的Al 原子。A 代表模型A ,B 代表模型B
对比模型A 与B 的吸附过程,虽然模型B 中AlN 分子中的N 距基片表面Al ②较远,仅仅是经历了较长的物理迁移与吸附过程,同样是N 、Al 先后分别与表面临近的Al 、O 成键。在稳定的吸附位置处, (Al) N - Al 键长为01172nm ,振幅为±01015nm ,吸附后AlN 的键长为01189nm ,振幅为±01010nm。吸附后的键长与吸附能表明AlN 在a2A1203 (0001)表面发生了较强的化学吸附。Al 在表面稳定的化学吸附位置S5、S7 正好偏离表面O 六角对称约30°,从图3A2 ④可以看出,吸附后AlN 平行于[2110 ] ,同表面Al ②的一个Al —O 键( [1010 ]方向) 有30°的偏转角度;同样在图3B2 ④中,吸附后AlN[12 10 ]与表面Al ②的另一个Al —O 键( [01 10 ]方向) 有30°的偏转角度。
在异质外延薄膜生长过程中,最初的外延取决定了薄膜的生长朝向,基底表面原子和最初吸附物的表面动力学过程对薄膜最终平面生长起到重要作用。因此,AlN 在a2Al2O3 表面的最初吸附决定了AlN 的成核与进一步薄膜的形成。通过两种吸附模型(模型A、模型B) 计算表明,当Al —O(基片) 成键后,在这些偏离基片表面O 位置30°处,是Al 较强的化学吸附位,所形成的Al - O 键能最大。从微观机制来看,化学键最强的方向应是晶体生长最快的方向,有利于薄膜小岛初期的形成。这样,随着AlN 成核与薄膜形成,在30°旋转方位处的原子排列有利于最初单层AlN 与Al2O3 亚晶格的调整,这将使其界面结构稳定,使得AlN 与蓝宝石之间的晶格失配度降低。
3、总结
通过模型A 与B 的动力学过程计算分析,AlN在α-Al2O3 (0001) 表面吸附过程为物理吸附与表面迁移、(Al)N - Al 化学吸附、Al - O(基片) 化学吸附、和最优吸附位置成键4 个阶段,AlN 分子在α-Al2O3(0001) 表面的结合能大于4.844eV。吸附后AlN 分子化学键与最近邻的表面Al - O 键有30°的偏转角度,Al 在表面较稳定的化学吸附位置S5、S7 正好偏离表面O 六角对称约30°,并减小甚至消除了吸附前表面Al - O 层的驰豫。