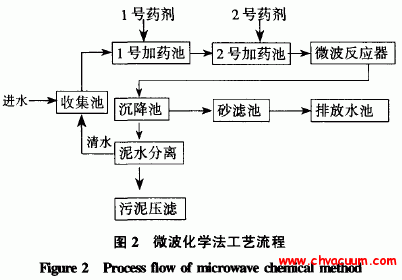
有机分子在金属表面上结构和动力学行为的第一性原理计算研究
有机分子在金属表面上结构和动力学行为的第一性原理计算研究
杜世萱
中国科学院物理研究所,北京市海淀区中关村南三街8 号,邮编100190
分子-金属体系具有独特的物理化学性质,在基础研究以及未来电子器件等应用领域受到人们越来越多的关注。金属酞菁化合物由于其热力学、化学稳定性及优良的光电特性等,在染料工业、光电功能材料等方面被广泛应用,同时,大量的实验工作也对该类分子的自组装结构和单分子物性等在基础研究层面进行了深入的研究。
我们利用基于密度泛函理论的第一性原理计算方法,系统地研究了金属酞菁,MPc(M = Mn, Fe, Co, Ni, Cu, Zn),分子体系在Au(111)面上的吸附构型、界面电子结构以及磁性过渡金属酞菁化合物的磁性性质等,与自由的金属酞菁分子的以上性质进行了比较分析,从本质上对酞菁分子在金属表面表现出来的的自组装行为及单分子物性给出了合理的解释。
研究结果表明,MnPc,FePc 和CoPc 分子与Au(111)的相互作用比NiPc, CuPc, ZnPc 与Au(111)的相互作用强;除CoPc 分子外,其他磁性金属酞菁分子吸附前后中心金属原子的磁矩保持不变;金属酞菁分子在Au(111)表面的吸附能都不大,并且不同构型间的能量差异较小。
这些研究结果不仅成功地解释了酞菁分子在Au(111)表面的自组装行为以及单分子物理性质,结合实验给出了动力学过程中分子亚稳态的构型,还对金属酞菁分子在Au(111)基底上的动力学行为给出了合理的解释,如,由于金属酞菁分子不同吸附构型间能量差异小,使得在特定温度下金属酞菁分子在Au(111)面上容易扩散,与实验上观察到的FePc 分子在液氮温度下仍然扩散的现象一致等。
参考文献:
(17) Y.Y. Zhang, S.X. Du, H.-J. Gao, Phys. Rev. B, 2011, 84, 125446
(18) N. Jiang, et al. Nano Lett., 2010, 10, 1184-1188
(19) Q. Liu, et al. Phys. Rev. Lett., 2010, 104, 166101
(20) L.Z. Zhang, et al. J. Phys. Chem. C, 2011, 115, 10791-10796.
(21) L. Gao, et al. Phys. Rev. Lett., 2008, 101, 197209
(22) L. Gao, et al. Phys. Rev. Lett., 2007, 99 , 106402.